

In 2020, governments around the world imposed brutal lockdowns across the population, promising a safe and effective Covid-19 vaccine was just around the corner.
The Trump administration made no secret of the fact that it wanted the Food and Drug Administration (FDA) to hurry the process along.
Stephen Hahn, then FDA commissioner, was summoned to the White House and asked to explain why he hadn’t moved faster to approve Pfizer’s Covid-19 vaccine.
It drew widespread criticism but Kayleigh McEnany, then White House press secretary, defended Trump saying he would “never apologize” for putting a fire under the FDA.
Hahn appeared to resist the pressure – publicly at least.
“We will make sure that our scientists take the time they need to make an appropriate decision,” said Hahn “It is our job to get this right and make the correct decision regarding vaccine safety and efficacy.”

Soon after the November 3, 2020 election, Pfizer’s vaccine authorisation was imminent. But Trump would soon be replaced by President-elect Joe Biden, and lamented that his successor would get all the credit.
“They will try and say that Biden came up with the vaccines,” said Trump to Fox News a week after the election. “The vaccines were me, and I pushed people harder than they’ve ever been pushed before.”
{kind=link}
Warp Speed Authorisation
Pfizer (and its partner BioNTech) submitted its trove of clinical trial data to the FDA on November 20, 2020.
The FDA completed its review and granted emergency authorisation for Pfizer’s investigational mRNA vaccine on December 11, 2020.
The entire process took only 22 days.
The FDA assured the public that it conducted “a rigorous review of laboratory and clinical data” but vaccine hesitancy was already sky-high.
Pew research found that 49% of Americans said they would ‘probably not’ or ‘definitely not’ get the vaccine in light of the fast-paced process.
Reviewing the Trial Data
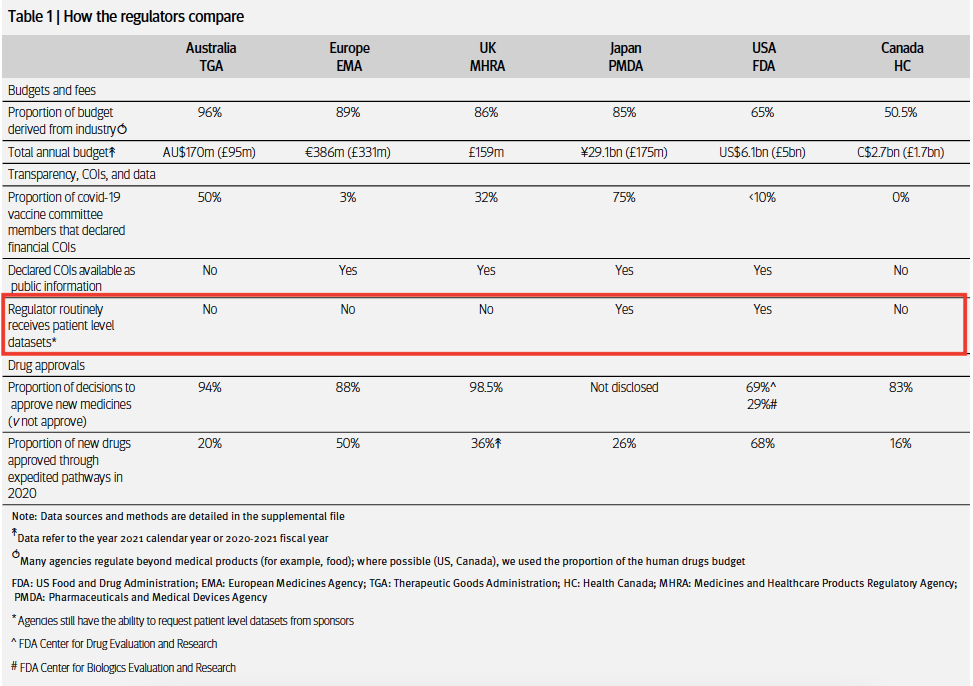
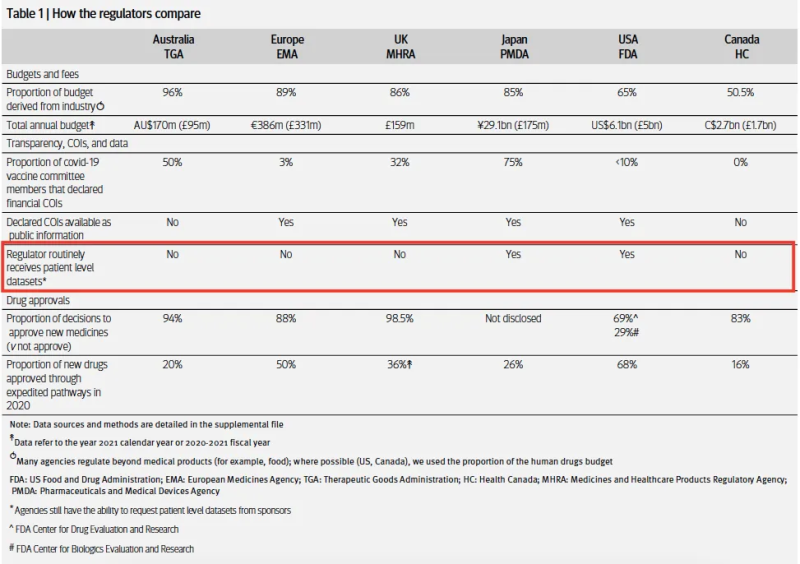
Most regulators review data that have been curated by the trial sponsor. Australia’s TGA for example, never received the source data from the trial, only the aggregated data from the manufacturer in a dossier.
The FDA, however, is one of the few regulators that receives and reviews the individual participant data (IPD) and other regulatory documents that underpin approval decisions.
The data sets are enormous.
Pfizer’s mRNA trial, for example, would collect data on each of the 44,000 subjects – consent forms, case report forms, baseline values, and tests from multiple follow-up visits.
Typically, a trial that size would gather hundreds of thousands, if not millions, of pages.
So, did the FDA receive the IPD and review it all?
The FDA told me that it did, in fact, receive the IPD for Pfizer’s mRNA trial, and in 22 days it “carried out a full analysis of those data prior to authorizing the vaccine for emergency use.”
The FDA’s vaccine chief Peter Marks boasted that the effort was ‘heroic.’
“This was not business as usual,” Marks wrote in STAT. “FDA undertook an all-hands-on-deck approach to this work.”
{kind=link}

Peter Gøtzsche is a Danish physician and has many years of experience reviewing regulatory documents. He ridiculed the idea that a “thorough” review could be completed in that timeframe.
“There’s no way the FDA carried out a ‘full analysis’ of the IPD in only 22 days,” remarked Gøtzsche. “It’s impossible. It would take a minimum of 6 months to pull off such a complex analysis.”
“There can be millions of pages to a single trial. And if you are going to do a thorough analysis, it requires very careful detective work to review it all,” he said.
{kind=link}

Gøtzsche, who authored the book “Deadly Medicines and Organised Crime” has been an expert witness in court cases for people who’ve been harmed by psychiatric drugs.
He says after analysing trial data that have been subpoenaed in legal cases, you really get a sense of just how much drug companies “lie and cheat” in the volumes of data.
“I know from experience that drug companies try to bury harms. For example, they might use different words to describe the same harm so that it doesn’t get picked up when you’re searching for those key terms in the documents,” said Gøtzsche.
“The only way the FDA could have finished a full analysis of Pfizer’s IPD in 22 days is if they cut corners or they only analysed the aggregated information submitted in the dossier,” he added.
FDA Denies it Cut Corners
The FDA rejected the idea that 22 days was insufficient time to thoroughly analyse the IPD.
“The FDA uses a team-based approach to evaluate and analyze data, and staff with relevant knowledge, beyond the immediate review team, are also involved,” stated the agency.
The FDA would not confirm exactly how many staff it assigned to the task, but added, “Review of clinical trial participant data generally involves staff from multiple offices in the center, who bring their considerable medical and scientific expertise to the process.”
But Gøtzsche said this is not the way it works when analysing regulatory filings.
“You can’t just divide up the pages among many staff members to get the work done quicker,” explained Gøtzsche.
“You need consistency to do the job right. It requires the same person reading the data and looking for patterns. The more pages that person analyses, the more familiar he or she becomes with how the data have been documented and what has been hidden,” added Gøtzsche.
More recently, regulatory documents have been digitised and are available electronically, meaning they don’t come in hard copy, stacks of paper.
In some instances, this enables researchers to perform ‘keyword searches’ to look for information, but sometimes, the pages have been scanned or photographed, which does not allow word searches.
My 2022 BMJ investigation found Japan’s drug regulator PMDA is the only other major regulator that routinely receives IPD.
{kind=link}
Currently, Tom Jefferson and colleagues at Trust the Evidence have spent the last few months exploring the regulatory data sets for licensure of Pfizer’s vaccine (Cominarty) thanks to a lawsuit filed against the FDA by Aaron Siri, US attorney acting on behalf of the non-profit group, Public Health and Medical Professionals for Transparency.
{kind=link}
Republished from the author’s Substack
Disclaimer
Some of the posts we share are controversial and we do not necessarily agree with them in the whole extend. Sometimes we agree with the content or part of it but we do not agree with the narration or language. Nevertheless we find them somehow interesting, valuable and/or informative or we share them, because we strongly believe in freedom of speech, free press and journalism. We strongly encourage you to have a critical approach to all the content, do your own research and analysis to build your own opinion.
We would be glad to have your feedback.

Source: Brownstone Institute Read the original article here: https://brownstone.org/