


Over the past decades of my career, I have spent countless hours working to protect Americans by researching the safety of drugs. My education and career have taken me through about half a dozen universities, Big Pharma, and at the FDA under three presidential administrations. Drug safety considers why one individual may take a pharmaceutical product and have zero adverse events, while a different individual could take the same product but have adverse reactions up to and including permanent disability or death. By default, studying drug safety also considers non-clinical aspects of manufacturing and drug quality as well.
Because drug quality is an essential factor in assessing drug safety, my trek to protect Americans led to my conceptualizing and founding of the world’s first “analytical pharmacy” missioned with scientifically verifying pharmaceutical products from places like India and China before dispensing them to patients. Unfortunately, the pursuit of largess over ethics and protecting patients led to that company’s financial management committing extensive FDA violations and being accused by judges of making false scientific claims (all of which uncoincidentally occurred following my exit).
Without outside confirmation of drug quality, Americans are completely dependent on the FDA and manufacturers to assess and confirm product purity. Drug safety has been shown to be a noteworthy problem when it comes to Covid mRNA injections. Unfortunately, if anyone wanted to conduct their own analysis on mRNA injections, they do not have an appropriately detailed ingredient list to compare it to, or even access to the established regulatory methodology on how to properly test it for purity.
It’s because manufacturers and the FDA consider all ingredients of these mRNA injections, including the sequence of mRNA plus lipid nanoparticle (LNP) properties including half-life, LNP structures, surface modification(s), number/type(s) of LNPs per dose and attachment points on the mRNA strand, to be unspecified or “trade secret.”
On top of that, the FDA additionally considers the methodologies on how to test mRNA injections for purity a trade secret as well.
Bipartisan Support and Hundreds of Billions of Taxpayer Dollars, but NO Transparency?
Covid mRNA secrecy exists even though both the Trump and Biden administrations had proposed full transparency with mRNA injections to the point of lifting Covid mRNA intellectual property rights. Despite that, both the FDA and manufacturers are permitting/keeping tight grips on patents, including basic data about these shots, as a trade secret. They are doing so despite all Covid vaccine manufacturers having received hundreds of millions of taxpayer dollars according to Forbes/Statista publications.
Studying Drug Safety Epidemiology is Difficult Enough. Without Verifiable Product Purity/Consistency, a Complete Safety Evaluation is Impossible.
Full transparency of all ingredients and quality control measures are important not only because they were heavily taxpayer-funded with hundreds of millions of dollars, but because a slew of questions have arisen about the safety and efficacy of Covid mRNA injections.
In addition to being exceptionally complex, their approval was expedited by regulators after less than one year. Most drugs and vaccines typically take around ten years to fully test for safety/efficacy and review and approve. In addition to the ingredients being completely novel, very complex, and the first of its kind to be administered on a massive scale, development including long-term clinical safety/toxicity evaluations and epidemiology reviews were expedited and likely not fully elucidated prior to release.
FDA Ingredient Verification, Transparency, and “Truthfulness” Have Precedents Dating Back to the 1800s:
The analytical verification and the transparency of ingredients or “truth in labeling” where the contents of the bottle are required to match the listed ingredients predates the establishment of the FDA, back to 1862. Today’s FDA was actually born out of what started off as a single “Department of Chemistry” employee employed at the US Department of Agriculture.
Adulteration, (altered or toxic ingredients) misbranding (contains a false label or is otherwise misleading, or contains incorrect medical claims), or mislabeling (contains ingredient(s) are not listed on a product label) have all had long, ugly histories in America. It was thought that the egregiousness had peaked in the early to middle 19th century – or at least that’s when it became identifiable – as only by 1862 had technical processes been developed to analyze and detect ingredient fraud. Prior to that, so-called “traveling medicine men” calling themselves “doctors” (invariably with dubious or nonexistent credentials) would peddle bottles of “cure-all” products, the ingredient labels of which would only list nebulous or innocuous contents such as “vitamins” “herbal extracts,” or “snake oil” – or often have no ingredient list at all.
Back then, many devout, puritan New Englanders, who for religious reasons would never touch alcohol, would purchase these solutions from these peddling hucksters and unknowingly be duped into consuming solutions which not only contained alcohol, but narcotics such as opium and/or cocaine. Under the pretense of improving a preposterously broad cornucopia of ailments, patients instead developed punishing addiction and/or otherwise had their health negatively impacted by these early “drug dealers.”
As the problem grew, the federal government began taking notice. Eventually, the Pure Food and Drug Act was passed in 1906 and led to the creation of the Food and Drug Administration (FDA).
[FDA had a formative duty of assuring that drugs bear truthful labeling statements and meet certain standards for purity and strength.
Remember that nearly 120-year-old truthful labeling requirement and “purity” portion of the Pure Food and Drug Act of 1906 as you read on about mRNA verification testing and ingredient transparency.]
What “Truthful” and “Pure” Ingredient Verification Testing is Occurring for FDA-Regulated Products?
Back in 2021, the FDA opted to start monitoring America’s pharmaceutical quality via a remote collection of mailed-in submission of samples for drugs as a substitute for live facility inspections because of the Covid pandemic. Was that legal? Could that ever be considered scientifically appropriate? Today, despite the pandemic having ended, the only official pharmaceutical release testing currently being performed on any Covid mRNA pharmaceutical appears to still be done by FDA via a manufacturer-supplied, “mailed-in” sample according to a screenshot of the current FDA website. Obviously, a “mailed-in” sampling method is far different and potentially less reliable than directly collecting samples via a direct, in-person collection method. Despite that, the FDA claims that it has “the highest standard across the globe for sampling and testing.”
Furthermore, the FDA is proposing further advancing its “mailed-in” remote testing policy with a newly proposed guidance document.
Although it only exists as a “draft” FDA document, official FDA websites show that the mailing in of samples appears to have already been implemented since at least January of 2021. The FDA appears to be asserting the results of those mailed-in tests as their independent verification.
Furthermore, the bottom of the first page of the FDA draft document proposes expansion of “remote testing.” It currently lists every FDA product-regulating division at the FDA, implying that it is an agencywide policy proposal.
The complete list includes the:
- Office of Regulatory Affairs
- Office of Food Policy and Response
- Office of Combination Products
- Center for Biologics Evaluation and Research
- Center for Drug Evaluation and Research
- Center for Devices and Radiological Health
- Center for Food Safety and Applied Nutrition
- Center for Tobacco Products
- Center for Veterinary Medicine
Is “Mailed in” Quality Control Sampling by the FDA Appropriate? What if States’ Health Department Restaurant Inspections Mirrored FDA Policy?
This “mail-in” sampling methodology is similarly absurd, for example, to a states’ health department monitoring restaurants by asking them to periodically “mail in” various items from their menu to a testing facility so that health departments can test for potential food-born contamination, and/or asking restaurants to promise to test menu items themselves. What if that restaurant was in China? What if that restaurant was in India? Or any other country well-known to have an abysmal history of fraud and quality control problems?
That methodology would be unacceptable for both restaurants and pharmaceutical companies, for reasons which include the obvious: manufacturers could send in the samples they prefer – not necessarily representative batch samples. It’s obviously not the same as FDA inspectors acquiring samples during unannounced inspections of the entire facility.
Under the restaurant analogy, of course all restaurants would submit “A” grade samples which would not necessarily be representative of what consumers receive.

Quality Control: What is Pharmaceutical “Release Testing” and Why is it Important?
Today, the FDA oversees the quality and content of $2.7 trillion worth of product annually, but seems to be suppressing critical ingredient verification assessments and results. The FDA is supposed to protect Americans by conducting comprehensive analytical testing as a checksum to assure ingredient accuracy. The results of that ought to be transparent to taxpayers which fund the FDA’s $6.6 billion budget. That scientific verification is referred to as pharmaceutical “release testing.” Release testing is a technical term referring to a process involving a variety of instrumentational analyses used to comprehensively test products for purity, concentration, consistency, identity, and impurities of any kind.
The entire FDA was born from that one “Department of Chemistry” employee from 1862 and the need for transparency and verification of ingredients. Today, that employee has proliferated into an entire FDA department of 1,300 scientists and support staff putatively dedicated to ingredient verification via pharmaceutical release testing. The FDA’s Office of Pharmaceutical Quality (OPQ) is supposed to make sure that pharmaceuticals exactly match the contents of the listed ingredients, without quality/impurity (qualitative) or content (qualitative) variability. The rules requiring that are very specific and detailed in 21 CFR § 201.10.
How the FDA Verifies mRNA Injections for Quality Control:
The quality control results from tests from mRNA injections were particularly critical because they are large, complex, and were rapidly made. While taxpayers depend on the FDA for verifying mRNA injection quality and sharing the results, the FDA seems obliged to protecting manufacturers’ ingredients at the expense of even the most basic transparency regarding mRNA Covid products. While the FDA seems to be collecting samples, their “mail-in” methodology is fundamentally flawed. Additionally, the FDA is not sharing the results of those tests anywhere I could locate them.
In other words: during the pandemic when brand new, broadly implemented mRNA shots were being thrust upon Americans at “warp speed” and when America was relying on the FDA’s quality/regulatory duties the most, the FDA was accepting self-submitted “mailed-in” quality control testing and/or results. Did the FDA not consider that mRNA manufacturers admitted that they “struggle[d]” to respond to manufacturing and were “scrambling” to keep up with manufacturing processes? Manufacturers of mRNA ingredients further stated that efforts to meet needs were “unprecedented.”
Statements like this do not yield consumer confidence in quality, and are illustrative of tremendous upscaling of these complex products that ought to warrant especially vigilant and in-person FDA scrutiny of facilities and manufactured products, pandemic or not. One mRNA ingredient manufacturer, for instance, stated that they suddenly ramped up their production by 50 fold.
In the midst of that novel technology pushed through at “warp speed” haste, were none of the 1,300 OPQ scientists at the FDA demanding live inspections, or at least, offering to do anything other than asking for potentially questionable “mailed-in” samples for testing?
The obvious question is: why didn’t the FDA collect samples directly? Even with the pandemic in place, the FDA could have inspected facilities wearing hazmat suits or – or at the very least – opted to collect samples from pharmacies, hospitals, or at distributor warehouses.
Concealed Methodology for Testing mRNA Injection Ingredients:
Beyond the absence of testing results and questionable “mailed-in” sampling results – the FDA is additionally concealing their validated methodology preventing others from performing their own, independent analyses on the quality/purity of mRNA injections.
Independently analyzing drugs for purity and potential contamination as compared to the ingredient list is something I had attempted to do myself when I conceptualized the world’s first analytical pharmacy. However, since mRNA shots are a novel technology with a less-than-fully-transparent ingredient list, the testing methodology one would need to employ is not straightforward as it would be for other small-molecule drugs. Anyone trying to look up the storage, stability, specificity, chemistry, sensitivity, or even basic methodology for testing validation and/or results are blockaded via an FDA report containing ludicrously invasive redactions, making even the most fundamental scientific comprehension of how to potentially evaluate results or conduct testing impossible.
As a poignant visual example, a single redacted page in a longer FDA regulatory summary (shown below) is part of a 127-page document (only 63 pages of which have been shared, and of those 63 pages, around 50% has been redacted) on how to evaluate the purity, concentration, and other analytical measures of mRNA injections.
Those FDA (b)(4) redactions specified detailed redactions used to “protect[s] trade secrets and confidential commercial or financial information.” But is it really appropriate to label it “commercial” if the research/development/product was funded with hundreds of millions of taxpayer dollars?

Without a list of ingredients or testing methodology, it is impossible for anyone else outside the FDA or manufacturers to know precisely how to check for product adulteration (altered or toxic ingredients) or mislabeling (because a full list of ingredient(s) including the nucleotide sequence and the lipid nanoparticle configurations are particularly vague on the product label).
The lack of methodology is particularly troublesome since new, preliminary data using independent methodology has shown evidence of DNA contamination in mRNA Covid injections.
So, if an outside individual claimed to have tested and found an impurity in mRNA shots and asked the FDA or manufacturers for its response, they would be met with some response stating something along the lines of:
- You did not use validated/appropriate testing methodology to come to your conclusions and therefore your analyses are invalid.
To that, the independent laboratory would attempt to request the testing methodology from FDA-approved documentation (ie, the full document containing Figure 5) by asking: “Okay, I would like to test it using your approved methodology; will you tell us what that is?”
- The FDA or manufacturer would reply something along the lines of: “What we are willing to disclose about the methodology employed that is not confidential may be found online, or via an FDA FOIA request” …where they would be met with the following heavily redacted document, where anything remotely meaningful is blanketed with (b)(4) redactions.
Reading between the lines: It’s obvious that both manufacturers and America’s FDA do not want anyone other than themselves to know the complete ingredients of or even test mRNA injections for purity and consistency.
According to FDA Officials: Pharmaceutical Manufacturing is Highly Prone to Error:
Many things can – and do – go wrong during the pharmaceutical manufacturing process. Beyond potential inconsistencies with mRNA/LNP injections, qualitative and quantitative issues implicate every FDA-regulated pharmaceutical product. Even the House and Senate have formally acknowledged reports of the FDA’s failure to secure America’s pharmaceutical supply chain. The majority of America’s pharmaceutical consumer-end-user products being produced overseas in countries like India and China, and other low-labor-cost countries are not well regarded for high levels of quality control. The Federal Register is riddled with reports of violations at Indian and Chinese manufacturing plants.
Is the FDA also certifying these plants – including those with long histories of violations – via a “mail-in” system to the FDA? Outrageously, the answer to the question is something that would make anyone concerned with pharmaceutical quality very uncomfortable.
While a Six Sigma precision level has long been the target for quality and safety in the automobile, computer, mobile telephone, and other high-tech manufacturing, it seems to have been mostly overlooked when it comes to pharmaceutical manufacturing.
FDA officials have published data estimating a of 2-3σ (sigma) of imprecision in pharmaceutical manufacturing. A 2σ quality corresponds to 308,537 defects per 1,000,000 opportunities. (There are likely a lot more than 1,000,000 opportunities for error when it comes to pharmaceutical manufacturing.) The FDA is aware of this at the highest levels of leadership; in fact, the current FDA’s head of the Office of Pharmaceutical Quality, Michael Kopcha even wrote and published the above Six Sigma calculation, lamenting the imprecise nature of pharmaceutical manufacturing back in 2017.
The latitude of error for mRNA products and/or their LNPs could be even less precise than the 2-3σ, (the lower the σ, the more erroneous a product is) since they include nucleotide material and novel LNPs, making them substantially more complex than small-molecule pharmaceuticals — notwithstanding their being developed, manufactured and released at “warp speed.”
With even the FDA and its officials recognizing an inherent manufacturing imprecision, why in the wide world of sports is the FDA not fulfilling its safety mission by publicly sharing its release testing of mRNA technology with the American public that funds them?
Pre-1862 Again? Are mRNA Shots the Only Drugs for Which Americans Don’t Have Complete Ingredient Information?
The lack of clarity on the number of sequences of mRNA shots and other critical information is in direct contrast to another FDA-approved RNA-based drug – patisiran (Onpattro®). Onpattro transparently provides the sequence, molecular weight, and milligram strength of its products within official FDA package labeling as illustrated in an excerpt below:


Lack of Covid mRNA Dose Specificity: 0.3mL (or 0.5mL) of What?
As of now, we still don’t have basic ingredient information on any Covid mRNA injection. Pharmacists only know to give a specific volume of fluid, and seemingly did so without question. Normally, official FDA package labeling should detail the actual ingredients in that volume, but not for Covid mRNA labels: they simply state 0.3mL (or 0.5mL) as the “Dosage Form and Strength.”
Additionally, as any high school student could tell you, 0.3/0.5mL is a volume, not a strength. We don’t know any quantitative specifics of what is contained in that 0.3/0.5mL such as: How many LNP particles? What size/morphologies of those LNPs? How many mRNA sequences in that volume?




Is This What Passes as Adequately Transparent or “Truthful Labeling” by the FDA?
The above cut-and-paste excerpt from the package insert is all of the information manufacturers are sharing with consumers regarding the dose – which is woefully inadequate as compared to all other FDA labels – or anyone who is curious to know anything beyond how much fluid to inject and the 30 or 100mcg concentration of an unspecified mRNA sequence.
The remarkable imprecision of this label permitted by the FDA seems to conflict with its nearly 120-year-old label specifically: “requiring that food and drugs bear truthful labeling statements and meet certain standards for purity and strength.”
Is this what passes as a “truthful” list of ingredients by the FDA? (See 21CFR §352, and 21 CFR §201.10 regarding “statement of ingredients” and “misbranded drugs and devices.”)
The question is: does listing unknown or nonspecific ingredients that nobody except the manufacturer can decipher really meet the spirit or legal requirements of “labeling?” Is that label what is considered “truthful” by America’s FDA? Whose side is the FDA on anyway; manufacturers or consumers?
In addition to it not being directly specified, the exact number of LNP nor mRNA strands in a 30 or 100mcg injection can’t even be extrapolated stoichiometrically or on the basis of Avogadro’s number, because the mRNA sequence, molecular weight, and/or LNP component/configurations aren’t provided anywhere within the official FDA labeling.
How can anyone know if the number of mRNA strands to encode the spike protein for Covid is proportional to the load of Covid inoculum that one would receive from a community acquired infection? Answer: they can’t.
Are Covid mRNA Injections Appropriately Labeled/Mislabeled?
21 CFR 211.125 specifies “Strict control shall be exercised over labeling issued for use in drug product labeling operations,” but it appears the FDA was so lax with its approved labeling of Covid mRNA injections despite the fact that every other drug – including mRNA-based Onpattro – does specify that information. Historically, FDA regulatory decisions (such as which information to include in product labeling) are based on precedence, and Covid mRNA shots were an obvious deviation from the FDA’s historical and legal precedence. That noteworthy data absence and lack of clarity sort of harkens back to the days of Morley’s Liver and Kidney Cordial in the late 1800s. The difference is: back then, the FDA didn’t exist, but today there is an FDA with ~20,000 employees, at least some of whom ostensibly believed that this label was transparent and “truthful.”
Stating an unknown/indecipherable/obscure ingredient that nobody could ever accurately determine likely isn’t what 1906 Pure Food and Drug Act lawmakers intended when they specified the FDA rules on “truthful labeling.” Separate from that: the fact that the doses are doubled per volume from different manufacturers (30mcg/0.3mL vs 100mcg/0.5mL) means that these mRNA sequences appear to be vastly different in nucleotide length, and in turn, would have more and different LNPs plus attachments. Does that mean that mRNA sequences used to transcribe the spike protein are around double the size (10mcg/0.1mL versus 20mcg/0.1mL) as compared to different manufacturers, or is something else contributing to the nucleotide length difference?
For the layman still reading up to this point (kudos, by the way): The lack of detailed labeling information could be like broadly advertising a house for sale, stating it is made of wood and bricks, on a cement slab – but not showing any pictures of the house, (e.g. sequence) and not sharing its square footage (e.g. molecular weight). In any case, the lack of information is inadequate and a deviation of traditional standards.
Every other FDA-approved drug — including other mRNA drugs — contains full ingredient disclosures on their products, including a structural representation and molecular weight of their product so people know exactly what they are getting.
It’s true: Look up whatever drug you can think of in the Drugs.com database and notice how all labels provide structure and/or molecular weight. Proof that Covid mRNA shots are a conspicuous exception to the historical FDA approval practice and “truthful label” rule.
2023 Danish Study Details Significant Clinical Variability Between Batches of mRNA Covid-19 mRNA Injections:
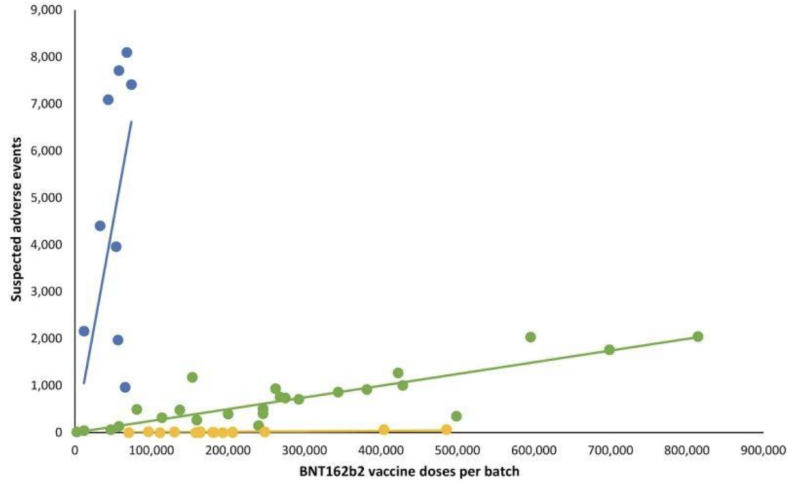
Not having any transparency on even potentially invalid “mailed-in” testing validation seems to have given manufacturers a pass on another critically important part of what the FDA oversees: potential clinical manifestations on lot/batch variations of mRNA shots. A retrospective Danish safety study published earlier in 2023 detailed a highly deviant pattern of adverse event reports from the Pfizer-BioNTech BNT162b2 mRNA injections as correlated with the Danish DKMA adverse event reporting system.
In the line graph which follows, different colored dots represent different batches of Pfizer-BioNTech’s mRNA injections. It separated out batches into three different categories; high- low- to (nearly) absent number of reported adverse event groups (blue, green, and yellow plots respectively).
In other words: putatively “equivalent” products from the same manufacturer seem to have wildly differing incidences of adverse events, by batch, with each of those batches representing hundreds of thousands of mRNA injections.
When corresponding linear regression lines were added, a particular pattern emerged:
Important questions about the noteworthy adverse events disparity between Covid-19 mRNA batches include:
- Could adverse event variances be due to qualitative or quantitative variances in mRNA sequences or number of mRNA strands between batches?
- Could adverse event variances be due to qualitative or quantitative variances in the size/morphologies or quantity of LNPs between batches? What tests have been performed to assure the safety of various LNPs used in mRNA injections?
- Were those batches that corresponded with the yellow versus green versus blue data points somehow qualitatively or quantitatively different?
- Was post-manufacturing storage/handling compromised at the administering facility (or somewhere else along the supply chain) leading to product variability?
- What is the Sigma/error rate of this and other products originating from the particular manufacturing facility/shift chief in charge of manufacturing?
- Were ingredients from these of Covid mRNA products sourced from India or China versus elsewhere, depending on the batch?
- What percentages of batches of Covid mRNA products were tested via in-person collection by an FDA inspector versus being “mailed in” from inception to date? Was every single batch tested using only either of these two collection methods?
- Did the FDA perform release testing verification on the Danish DKMA adverse event reporting system lots? If yes, why isn’t the FDA releasing those particular testing results? If not, why wasn’t testing done?
- Is there a fundamental problem with consistently producing LNPs and/or mRNA sequences reliably and without contamination?
The results of the Danish study and the above questions about adverse events could *begin* to be addressed, but not without the FDA independently sharing the results of their release testing findings. As it stands, because of ubiquitous FDA (b)(4) redactions, nobody knows the validated methodology for testing Covid mRNA shots or exactly which lots in the Danish study were or were not tested or the results of those batch tests.
…Then again, even if the FDA had chosen to release those batch test results, how do consumers know if those results are representative of batches specified, since manufacturers are self-selecting which samples to “mail in?”
Not providing ingredient transparency and assuring quality via an appropriate sampling methodology is a fundamental and basic requirement of the FDA. In fact, it was the primary reason for the formation of the FDA! Don’t Americans deserve better transparency, oversight, and “truthful labeling” laws when it comes to our pharmaceuticals – especially since those laws were made over 100 years ago?
Disclaimer
Some of the posts we share are controversial and we do not necessarily agree with them in the whole extend. Sometimes we agree with the content or part of it but we do not agree with the narration or language. Nevertheless we find them somehow interesting, valuable and/or informative or we share them, because we strongly believe in freedom of speech, free press and journalism. We strongly encourage you to have a critical approach to all the content, do your own research and analysis to build your own opinion.
We would be glad to have your feedback.

Source: Brownstone Institute Read the original article here: https://brownstone.org/